


|
|
|
|
DESCRIPCION La capecitabina es un derivado del 5-fluoruracilo (5-FU) que se puede administrar por vía oral, ocasionando unos niveles plasmáticos de 5-FU similares a los que se consiguen mediante una infusión continua de este último. Mecanismo de acción: al ser el principio activo de la capecitabina el 5-fluoruracilo, su mecanismo de acción es el de éste último. En resumen, este fármaco actúa interfiriendo con la síntesis del RNA y DNA por inhibición de la timidilato sintasa. Efectos sobre el RNA Para incorporarse al RNA, el fluoruracilo se debe transformar en monofosfato de fluoruracilo, lo que se puede producir de dos maneras:
Efectos sobre el DNA: el fluoruracilo puede ser transformado en fluordeoxiuridina mediante la acción de la timidina fosforilasa y luego a fluordeoxiuridina monofosfato a través de la timidina kinasa. Alternativamente, la fluordeoxiuridina monofosfato puede formarse indirectamente mediante la conversión de la fluoruridina difosfato a fluorodeoxiuridina difosfato y luego a fluorodeoxiuridina monofosfato. Esta ultima es capaz de formar un enlace covalente, firme pero reversible con la timidilato sintasa en presencia de metilentetrahidrofolato. La ligazón de la fluorodeoxiuridina monofosfato a la timidina sintasa bloquea la síntesis del timidinilato a partir del uracilo. Como el timidinilato es el precursor de la timidina trifosfato, uno de los cuatro deoxiribonucleótidos necesarios para la síntesis de DNA, su deficiencia ocasiona la depleción del trifosfato de timidina y, la interrupción de la síntesis de DNA. Además, tanto la fluorodeoxiuridina monofosfato como la fluorodeoxiuridina difosfato pueden ser convertidas a fluordeoxiuridina que puede ser incorporada al DNA mediante la DNA-polimerasa en lugar de la timidina trifosfato ocasionando un DNA aberrante. Además, cuando se administra en combinación con la leucovorina, los efectos del 5-fluoruracina son incrementados al estabilizarse el complejo timidilato sintasa-metilentetrahidrofolato-fluorodeoxiuridina Citotoxidad: durante las primeras 24 h de exposición al 5-fluoruracilo, se observa una citoxicidad en la fase S del ciclo celular, probablemente debida a los efectos del fármaco sobre el DNA. A las 24 horas, la citotoxidad tiene lugar en la fase G-1, probablemente a consecuencia de la incorporación del 5-FU en el RNA. La selectividad del 5-fluoruracilo hacia las células en división rápida se debe a que las concentraciones de timidilato sintasa son 20 veces mayores en las células en división que en las células no proliferantes.
|
|
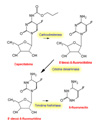 |
Farmacocinética: la capecitabina se absorbe rápidamente después de su administración oral siendo metaboliza en tres pasos al fármaco activo, el 5-fluoruracilo. En el hígado, el fármaco es hidrolizado mediante la acción de una carboxiesterasa a 5'-deoxi-5-fluorocitina, que es seguidamente transformada a 5'-deoxi-5-fluoruridina (*) mediante la acción de la citidina desaminasa, una enzima presente en números tejidos, incluyendo los tumorales. Finalmente, la enzima timidina fosforilasa hidroliza la 5'-deoxi-5-fluorouridina al fármaco activo, 5-fluoruracilo. Un factor que influye en la mejor actividad de la capecitabina en comparación con el 5-FU es que los tejidos tumorales expresan la timidina fosforilasa en mayor proporción que los tejidos normales, lo que ocasiona unas concentraciones más altas del antimetabolito en el tumor Toxicidad: en los estudios de toxicidad realizados con dosis múltiples, la administración oral diaria de capecitabina a macacos de Java y ratones se asoció a efectos tóxicos sobre los sistemas gastrointestinal, linfoide y hematopoyético, típico de las fluoropirimidinas. Estos efectos tóxicos fueron reversibles. Se ha observado con la capecitabina toxicidad cutánea, caracterizada por cambios degenerativos/regresivos. La capecitabina no causó toxicidad hepática ni sobre el SNC. Se ha detectado toxicidad cardiovascular (p.ej. prolongación de los intervalos PR y QT) en macacos de Java tras la administración intravenosa (100 mg/kg) pero no así tras la administración oral repetida (1379 mg/m2/día). En los estudios de carcinogenesis de dos años realizado en ratones no ha evidenciado carcinogenicidad con capecitabina. Durante los estudios de fertilidad estándar, se observó una alteración de la fertilidad en ratones hembras tratadas con capecitabina; sin embargo, este efecto revertió después de un descanso terapéutico. Además, durante un estudio de 13 semanas, aparecieron cambios degenerativos y atróficos en los órganos reproductores de los ratones macho; no obstante, estos efectos fueron reversibles después de un descanso terapéutico.En los estudios sobre embriotoxicidad y teratogenia efectuados en ratones se observó un incremento en las reabsorciones fetales y en la teratogenia que guardaba relación con la dosis. A altas dosis se observaron abortos y muertes embrionarias en los monos, pero ningún signo de teratogenia. La capecitabina no fue mutagénica in vitro para bacterias (test de Ames) o células de mamífero (ensayo de mutación génica V79/HPRT de hámster Chino). No obstante, como ocurre con otros análogos de los nucleósidos (ej: 5-FU), la capecitabina mostró efecto clastogénico sobre los linfocitos humanos (in vitro) y una tendencia positiva en los tests de micronúcleo de médula ósea murina (in vivo). INDICACIONES Y POSOLOGIA
La capecitabina está indicada como monoterapia en el tratamiento adyuvante de los pacientes que han experimentado una resección completa del tumor primario de colon tipo C de Dukes. Aunque el tratamiento con la capecitabina prolonga la supervivencia, sí que ha demostrado aumentar el tiempo libre de enfermedad en comparación con la combinación 5-fluoruracilo/leucovorina Administración oral
En los pacientes con cáncer colorectal metastatizado, la capecitabina en monoterapia es igual de efectiva que la combinación 5-fluoruracilo/leucovorina
En combinación con docetaxol en pacientes con cáncer de mama metastásico en el que haya fracasado un tratamiento previo con antraciclinas. Administración oral
- En combinación con una quimioterapia basada en platinos para tratar a pacientes con cáncer gástrico en fase avanzada Administración oral:
- En combinación con una quimioterapia basada en cisplatino + epirrubicina Administración oral:
Las dosis de capecitabina deberán ser individualizadas si se producen reacciones adversa. No se recomiendan modificaciones de las dosis para reacciones de grado 1 (véase CTCAE).La toxicidad debida a la administración de capecitabina se puede controlar mediante tratamiento sintomático, reducción de la dosis o interrupción del tratamiento. Una vez que se reduzca la dosis, no deberá incrementarse en ningún momento posterior. Se debe informar a los pacientes sobre la necesidad de interrumpir inmediatamente el tratamiento en caso de que se presente toxicidad moderada o grave. Insuficiencia hepática: se desconocen la seguridad y eficacia en pacientes con insuficiencia hepática. No se dispone de información relativa a insuficiencia hepática por cirrosis o hepatitis, aunque en principio, no parecen ser necesarios reajustes en las dosis Insuficiencia
renal: la capecitabina está contraindicada en pacientes con insuficiencia
renal grave (aclaramiento de creatinina basal por debajo de 30 ml/min.
La incidencia de las reacciones adversas grado 3 o 4 en pacientes con
insuficiencia renal moderada (aclaramiento de creatinina basal de 30-50
ml/min) está aumentada con respecto a la población general.
En los pacientes con insuficiencia renal moderada basal se recomienda
una reducción del 75% de la dosis inicial. En pacientes con insuficiencia
renal leve basal (ClCr 51-80 ml/min) no es preciso un ajuste de la dosis
inicial. Se recomienda realizar una cuidadosa monitorización
e interrumpir rápidamente el tratamiento si el paciente desarrolla
una reacción adversa grado 2, 3 o 4 durante el tratamiento. Estas
recomendaciones sobre ajuste posológico en caso de insuficiencia
renal rigen tanto para la Ancianos: No se necesita ajuste inicial de dosis de la capecitabina cuando se administra en monoterapia, aunque los ancianos muestran una mayor propensión a las reacciones adversas. Para los pacientes de 60 o más años se aconseja empezar el tratamiento con una reducción de dosis de capecitabina al 75% (950 mg/m2 dos veces al día) cuando se vaya a combinar este medicamento con docetaxel. Si no se observa toxicidad en pacientes ?60 años tratados con una dosis inicial reducida del fármaco en combinación con docetaxel, la dosis de capecitabina podría aumentarse con precaución a 1250 mg/m2 dos veces al día.
|
|
|
CONTRAINDICACIONES Y PRECAUCIONES La capecitabina está contraindicada en pacientes con alergia al fármaco o a cualquiera de los componentes de la formulación. Igualmente, está contraindicada en pacientes con hipersensibilidad conocida al 5-fluoruracilo y en pacientes con deficiencia de dihidropirimidina deshidrogenasa. También está contraindicada en pacientes con grave disfunción renal (aclaramiento de creatinina < 30 ml/min) La capecitabina está contraindicada en pacientes con leucopenia, neutropenia o trombocitopenia graves. Los pacientes tratados con capecitabina deberán ser controlados por un médico especialista en el manejo de los fármaco quimioterápicos. Los pacientes tratados concomitantemente con capecitabina y anticoagulantes cumarínicos deberán monitorizar con frecuencia el INR y el tiempo de protrombina, ajustando adecuadamente sus dosis de anticoagulante. La capecitabina aumenta de forma significativa el tiempo de protrombina y el INR, habiéndose producido casos de hemorragias, incluso fatales, en algunos pacientes anticoagulados a los pocos días de iniciarse un tratamiento con capecitabina. Incluso se han descrito casos de hemorragias al cabo de un mes de haber sido discontinuado el fármaco. Estas interacciones tienen lugar tanto en los pacientes con metástasis hepáticas como en pacientes en los que el hígado no está afectado. Los pacientes con más de 60 años e historia de cáncer están más predispuestos a experimentar este tipo de coagulopatía.
|
||
|
|
La capecitabina se clasifica dentro de la categoría C de riesgo en el embarazo. En los ratones, la administración de este fármaco en dosis de 198 mg/kg/día durante el período de organogenesis indujo malformaciones fetales y la muerte de los embriones. Estas dosis producen unos niveles plasmáticos de 5'-fluorodeoxiuridina 5 veces menores que los que se obtienen en los pacientes tratados con las dosis recomendadas de capecitabina. Las malformaciones observadas en los animales fueron fisura palatina, anoftalamia, microoftalmía, oligo- y polidactilia y dilatación de los ventrículos cerebrales. En los primates las dosis de 90 mg/kg/día administradas durante el período de organogenesis (dosis que ocasionan una AUC de la 5-'fluorodeoxiuridina un 60% inferior a la del ser humano) indujo la muerte fetal. No se han llevado a cabo estudios clínicos controlados sobre los efectos de la capecitabina en el embarazo. Si este fármaco se administra a mujeres fértiles, se les deberá advertir del riesgo que asumen si se quedan embarazadas
|
|
|
INTERACCIONES La administración de capecitabina con antiácidos aumenta ligeramente su absorción con un pequeño aumento de los niveles plasmáticos de 5'-deoxi-5-fluorocitina. Los otros metabolitos, incluyendo el 5-FU no son afectados. Se han comunicado algunos casos en pacientes estabilizados con fenitoína que mostraron un aumento de la toxicidad del anticonvulsivante cuando se inició un tratamiento con capecitabina. Aunque no se han llevado estudios específicos para determinar esta interacción, es muy probable que el aumento de la toxicidad se deba a la inhibición por parte de la capecitabina de las isoenzimas CYP 2C9 responsables de la biotransformación de la fenitoína. Como se ha indicado anteriormente existe una interacción entre la capecitabina y la warfarina. Un estudio en voluntarios ha puesto de manifiesta que una dosis única de capecitabina aumenta los niveles plasmáticos y la AUC de la warfarina de forma significativa. Los pacientes anticoagulados deberán chequear frecuentemente el IRN y el tiempo de protrombina y, probablemente, reducir las dosis de la cumarina. La leucovorina aumenta las concentraciones plasmáticas y la toxicidad potencial del 5-fluoruracilo. Se han descrito casos de graves e incluso fatales enterocolitis, diarrea y deshidratación en pacientes ancianos tratados con leucovorina y 5-FU. Aunque no se han realizado estudios clínicos específicos, se deberá tener en cuenta que la capecitabina puede afectar al metabolismo y aclaramiento de todos los fármacos que utilizan el sistema CYP 2C9 para su biotransformación Se desconoce si la presencia de alimentos afecta de alguna manera la farmacocinética de la capecitabina. El fabricante recomienda la administración del fármaco 30 minutos después de una comida debido a que la presencia de esta minimiza los efectos secundarios gastrointestinales de la capecitabina Se ha descrito una interacción clínicamente significativa entre la sorivudina y el 5-FU originada por la inhibición de la dihidropirimidina- deshidrogenasa por la sorivudina. Esta interacción, que provoca un aumento de la toxicidad de la fluoropirimidina, puede ser fatal. Por lo tanto, la capecitabina no debe administrarse con sorivudina o sus análogos químicamente relacionados, tal como la brivudina.
|
||
|
REACCIONES ADVERSAS Las reacciones adversas relacionadas con los tratamientos con capecitabina en monoterapia o en combinación con otros quimioterápicos mas frecuentemente observadas son las alteraciones gastrointestinales (especialmente diarrea, náuseas, vómitos, dolor abdominal, estomatitis), fatiga y síndrome mano-pie (eritrodisestesia palmo-plantar).
|
||
|
|
La eritrodisestesia palmo-plantar se presenta en el 57% de los pacientes. Un reciente estudio parece indicar que la aplicación de henna (colorante obtenido de la Lawsonia inermis) reduce los síntomas de este síndrome. | |
| Otras reacciones adversas sobre la piel son rash (7%), alopecia (6%), eritema (6%), sequedad de la piel (5%), prurito (2%), hiperpigmentación de la piel (2%), dermatitis (2%), rash macular (1%); descamación cutánea (1%), alteración de la pigmentación (1%), alteración ungueal (1%). | ||
|
|
Los trastornos digestivos más frecuentes son diarrea (47%), vómitos (18%), náuseas (35%), estomatitis (23%), y dolor abdominal (11%). Menos frecuentes son el estreñimiento (6%), dolor del tracto superior del abdomen (6%), dispepsia (5%), flatulencia (3%), sequedad de boca (3%), incontinencia fecal (2%), y hemorragia gastrointestinal (2%). | |
|
Las reacciones adversas sobre el estado general del paciente son fatiga (16%), astenia (10%) pirexia (6%), letargia (6%), edema periférico (3%), malestar (1%),y dolor torácico no cardíaco (1%) Sobre el sistema nervioso central se han descrito cefalea (4%), letargia (1%), vértigo (5%), parestesia (3%), y disgeusia (5%). Como reacciones adversas oculares se han descrito aumento del lagrimeo (5%), conjuntivitis (4%),e irritación ocular (1%) Finalmente, se han descrito hiperbilirrubinemia (3%) disnea (3%), epistaxis (2%), tos (1%), rinorrea (1%), dolor en las extremidades (3%), dolor de espalda (2%), artralgia (2%), disminución de peso (2%), y alteraciones en las pruebas de la función hepática (1%) Estas reacciones adversas oscilan entre el grado 1 y 4 de la CTCAE y pueden obligar a una reducción de las dosis o a la discontinuación del tratamiento.
|
||
|
|
PRESENTACIONES XELODA,
60 comprimidos de 150mg ROCHE
|
|
|
REFERENCIAS
|
||
|
||
Monografía creada el 15 de octubre de 2008.Equipo de redacci�n de IQB (Centro colaborador de La Administraci�n Nacional de Medicamentos, alimentos y Tecnolog�a M�dica -ANMAT - Argentina).
UN LIBRO IMPRESCINDIBLE
 |
||
|
|


