|
SINDROME
DE CHARCOT-MARIE-TOOTH
|
|
|
|
|
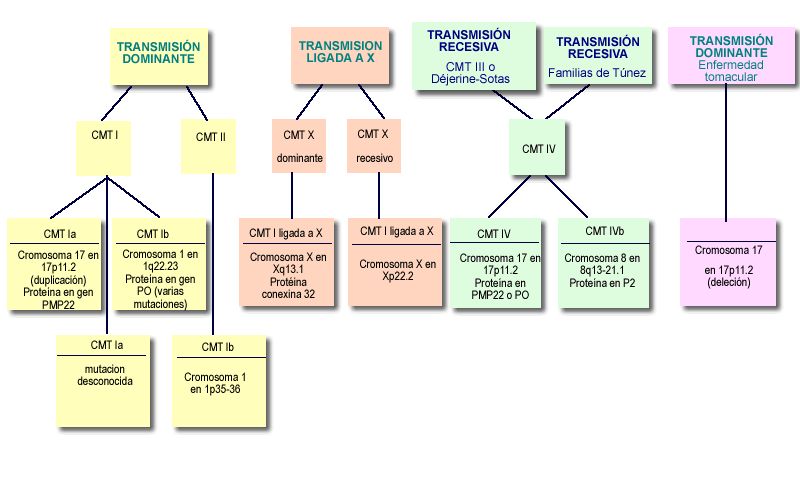
La enfermedad de Charcot-Marie-Tooth se clasifica en función de criterios genéticos, conociéndose varios tipos y subtipos CMT I y CMT II: comienzan usualmente en la niñez o adolescencia y son de caracter dominante autosómica. Son las dos formas más comunes de CMT. (De hecho, un subtipo de CMT I llamado CMT Ia, causado por un defecto en el gen PMP22 en el cromosoma 17, es responsable de cerca de un 60 por ciento de todos los casos de CMT). La CMT I es causada por la desmielinización y la CMT II es causada por axonopatía. La CMT II se asocia a veces con una condición tratable llamada síndrome de piernas inquietas, una necesidad irresistible de mover las piernas al estar sentado o acostado CMT III y CMT IV: también se inician en la infancia y son de carácter autosómico recesivo. La CMT III, más conocida como enfermedad de Dejerine-Sottas, se caracteriza por una desmielinización marcada por un inicio temprano y debilidad distal severa. Los niños con DS son lentos para llegar al desarrollo motor y a veces nunca logran la habilidad de caminar. Otros comienzan a caminar en la edad preescolar o más tarde, pero pueden requerir sillas de ruedas al llegar a la adolescencia. La escoliosis y la ataxia son comunes. (Algunos médicos consideran la DS una forma severa de CMT1, porque ambas enfermedades han sido conectadas con diferentes defectos en los mismos genes, incluyendo el PMP22). CMT X: la enfermedad de CMT ligada al cromosoma X tiene síntomas similares a los de CMT I y CMT II, pero a menudo afecta a los varones más severamente que a las mujeres. |