INSUFICIENCIA OVÁRICA PREMATURA [ICD-10: E28.3]
|
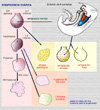
DEFINICIÓN El fallo ovárico prematuro (o insuficiencia ovárica precoz) es el cese patológico de la actividad ovárica antes de la edad normal de la menopausia, que hoy día se sitúa en las proximidades de los 50 años, con la consiguiente amenorrea, pérdida de la fertilidad y disminución de la secreción de estrógenos. En general, se admite como insuficiencia ovárica precoz el cese no fisiológico de la ovulación antes de los 40 años, siendo un problema relativamente frecuente que afecta aproximadamente al 1-4% de las mujeres en edad fértil. La condición se caracteriza por amenorrea, elevación de las gonadotropinas (> 40 mU/mL), y disminución de los estrógenos (< 20-50 pg/mL) El fallo ovárico prematuro es un síndrome producido por múltiples causas (*) que ocasionan la desaparición progresiva de los folículos ováricos o la falta de respuesta de estos a los estímulos normales. Muchas veces, el problema surge durante el desarrollo embrionario del ovario y otras, por factores externos. En condiciones normales, las gónadas empiezan a formarse cuando el embrión tiene unos 5 mm, a las 5-6 semanas de edad, a partir de un masa de células que forman crestas situadas medialmente en relación con los cuerpos de Müller y de Wolff, las crestas gonadales (*). Los ovocitos primarios (o células germinales) procedentes de la pared posterior del intestino primitivo, migran a los conductos mesonéfricos que constituirán, conjuntamente con las crestas gonadales, los ovarios embrionarios (*) . Las células germinales (también llamadas ovogonias u oogonias) son, a las 20 semanas de la gestación de unos 6-7 millones. Poco después, hacia la semana 25 comienza un proceso de atresia de estas células, de manera que al nacer, solo quedan en el ovario 1-2 millones de ovogonias. La atresia continua durante la infancia y pubertad y, al llegar la menarquia sólo quedan 300 o 400.000, de las cuales en cada ciclo menstrual unas 1.000 inician un desarrollo folicular y sólo una ovula, atresiándose las restantes (*) Algunos autores distinguen el fallo gonadal precoz (que se refiere a la reducción del número de folículos que tiene lugar durante la vida fetal y la infancia) del fallo ovárico prematuro que tiene lugar después de la menarquia. Otros, clasifican los distintos tipos de atresia y disfunción folicular dentro de un cuadro global de fallo ovárico prematuro (*) FACTORES ETIOLOGICOS Alteraciones cromosómicas Se observan alteraciones cromosómicas en el 20-50% de los fallos ováricos prematuros. Estas pueden ser tanto numéricas como estructurales. Numéricas:
Estructurales:
|
|
|
|
|
Alteraciones inmunológicas Frecuentemente, el fallo ovárico prematuro está asociado a enfermedades autoinmunes siendo las más frecuentes la tiroiditis autoinmune, el hipoparatiroidismo, la diabetes mellitus tipo 1, y la enfermedad de Addison. Sin embargo, otros muchos desórdenes autoinmunes han sido asociados al fallo ovárico prematuro. |
|
REFERENCIAS
|
|
| Monografía revisada el 5 de febrero de 2008. Equipo de Redacción de IQB |