|
 |
 |
SINDROME DE HURLER [ICD-10: E76.0]
|
|
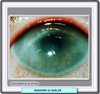
El síndrome de Hurler es un desorden de carácter autosómico recesivo que resulta de mutaciones en el gen que codifica la a-l-iduronidasa. Pertenece al grupo de enfermedades denominadas mucopolisacaridosis. La enfermedad se caracteriza por retraso mental, hepatoesplenomegalia, malformaciones esqueléticas y cardiopulmonares que producen usualmente la muerte en la primera década. Los individuos afectados parecen normales en el momento del nacimiento pero pronto muestran un crecimiento exagerado de los rasgos faciales, seguido de un retraso del crecimiento en general. En los primeros dos años, el diagnóstico es sugerido por la hepatoesplenomegalia, opacidades de las córneas (*) , facciones toscas y disostosis múltiples que se pueden observar en las radiografías. El retraso en el desarrollo es aparente al final del primer año de vida. Otros síntomas y signos adicionales que pueden acompañar esta enfermedad son sordera, infecciones respiratorias crónicas, enfermedad valvular cardíaca y aumento del tamaño de los ventrículos cerebrales, con retraso mental La manifestación dermatológica más frecuente del síndrome de Hurler es la hipertricosis generalizada. Algunos autores señalan el síndrome de Hurler como una enfermedad que puede darse en asociación con la dermatitis atópica Hasta hace muy poco, la enfermedad de Hurler no tenia tratamiento. Si la enfermedad es diagnosticada antes de que se hayan producido grandes lesiones y se encuentra un donante adecuado, el trasplante de médula ósea puede ser una opción. En algunos niños, el trasplante de médula ósea ha mejorado el curso de la enfermedad al producir las células trasplantades la enzima deficitaria En la actualidad, la terapia enzimática sustitutiva con iduronidasa obtenida por ingenieria genética (laronidasa) mejora notablemente todos los sintomas de esta enfermedad, así como la de Hurler-Scheie. Sin embargo, se desconocen los efectos a largo plazo de esta terapia, sobre todo a nivel neurológico. La mucopolisacaridosis I tiene el número de código 252800 de McKusick |
REFERENCIAS
|
|
|
|
| Monografía revisada el 10 de enero de 2005. Equipo de Redacción de IQB |