Riñón poliquístico [ICD-1o: Q61.3] |
|
|
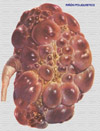
El riñón poliquístico es una condición hereditaria caracteriza por la presencia de numerosos quistes llenos de fluido que se desarrollan en los túbulos del riñón (*) pudiendo alcanzar un gran tamaño y peso. Es una de las enfermedades hereditarias más frecuentes (1:1000) y la causa genética más frecuente de las insuficiencias renales terminales. La enfermedad se produce por la dilatación progresiva bilateral de los túbulos renales para formar los quistes. También pueden ocurrir al mismo tiempo quistes hepáticos, aneurismas cerebrales y anormalidades de las válvulas cardíacas. Aunque se considera el riñón poliquístico como una enfermedad sistémica, en realidad menos del 5% de las nefronas se transforman en quistes. La enfermedad se debe a mutaciones en los genes PKD1 (que se encuentra en el locus 16p13.3-p13.12), PKD2 (4q21-q23) que codifican sendas proteínas la policistina-1 y la policistina-2. Un tercer gen, el PDK3 todavía no identificado también parece estar implicado en menor medida a esta enfermedad. La más frecuente, el riñón poliquístico tipo autosómico dominante, de debe a mutaciones en el PKD1. En la clasificación del riñón poliquístico intervienen los siguientes factores: causas (heredado o adquirido), características (único, múltiple, simple, complicado) y su localización (cortical o medular) (*) La morbilidad del riñón resulta de una disfunción renal progresiva que resulta de la invasión del parénquima renal por los quistes llegando hasta la enfermedad renal terminal. Aproximadamente la mitad de los pacientes con riñón poliquístico necesitan un trasplante renal antes de los 60 años. Ocasionalmente, algunos pacientes con riñón poliquístico pueden experimentar una hemorragia subaracnoidea como consecuencia de un aneurisma cerebral. Riñón poliquístico tipo autosómico recesivo: Se caracteriza por la asociación de quistes renales, fibrosis hepática congénita y, en ocasiones, dilatación no obstructiva de los conductos biliares intrahepáticos (enfermedad de Caroli). Los riñones son grandes y conservan su silueta típica. Los quistes son alargados, orientados radialmente de la médula a la corteza y de tamaño uniforme (*), ocupando la casi totalidad del parénquima. Va siempre acompañado de una fibrosis hepática (*) La intensidad de las manifestaciones renales depende de la edad de presentación. La forma perinatal cursa con riñones muy grandes (*) , muerte precoz por insuficiencia renal y fibrosis hepática mínima; la forma neonatal se presenta con hipertensión, uremia y escasa afección hepática; la forma infantil se manifiesta a partir de los 3 meses y cursa con manifestaciones renales y hepáticas (con posible progresión hepática tras un trasplante renal); la forma juvenil se presenta en adolescentes y en ella predomina la afección hepática, con hipertensión portal Las características ecográficas de los riñones incluyen el aumento de tamaño y de ecogenicidad (especialmente en la médula) y, a menudo, la presencia de pequeños quistes (< 2 cm) (*) La asociación constante de fibrosis hepática congénita determina que el aspecto ecográfico del hígado (aumento de tamaño y de ecogenicidad) y la biopsia hepática sean de utilidad en casos dudosos. El tratamiento consiste en un control estricto de la hipertensión arterial, tratamiento de las complicaciones metabólicas de la insuficiencia renal crónica y, eventualmente, diálisis y trasplante renal. Los enfermos con hipertensión portal son posibles candidatos para un shunt portosistémico. |
|
Riñón poliquístico tipo autosómico dominante Enfermedad multisistémica que afecta predominantemente el riñón y en grado variable otras vísceras (hígado, páncreas, bazo, etc.) así como los sistemas cardiovascular (aneurismas, enfermedad valvular), digestivo (diverticulosis) y musculosquelético (hernias). Los riñones suelen ser de gran tamaño y contienen numerosos quistes de dimensiones variables (*) en la corteza y en la médula. Histológicamente el epitelio que reviste los quistes presenta áreas hiperplásticas de tipo papilar o polipoide. Las membranas basales pueden ser normales o estar muy engrosadas o laminadas. Histológicamente los riñones poliquísticos muestran numerosos divertículos en todos los segmentos tubulares (*) . El mecanismo responsable de la formación de estas dilataciones focales de los túbulos renales podría ser una alteración de la membrana basal o del tejido conjuntivo o una proliferación primaria excesiva del epitelio tubular. Al aumentar de tamaño pierden su carácter tubular y crecen de forma autónoma, supuestamente por un proceso coordinado. Algunos factores ambientales pueden ejercer un efecto en la formación de los quistes Los quistes hepáticos se desarrollan por la dilatación progresiva de microhamartomas biliares, que están formados por pequeñas agrupaciones de conductos biliares rodeados por tejido fibroso. Los primeros síntomas pueden presentarse a cualquier edad, con mayor frecuencia entre la tercera y la quinta décadas de la vida. Consisten en dolor abdominal o lumbar, hematuria, hipertensión o, con menor frecuencia, infección urinaria, o presencia de una masa abdominal. Las causas de dolor abdominal se deben a la distensión producida por los quistes, hemorragia intraquística, hematuria macroscópica con coágulos, hematoma perinéfrico, nefrolitiasis o, raras veces, hipernefroma. También puede ser causado por quistes hepáticos con alguna complicación, diverticulitis y aneurismas aórticos. La hipertensión arterial a menudo se desarrolla cuando la función renal es todavía normal. A menudo, pero no siempre, los pacientes cuya poliquistosis renal autosómica dominante se ha detectado clínicamente y sobreviven el número de años suficiente desarrollan una insuficiencia renal. Entre el 5 y el 10% de los enfermos en programas de diálisis y trasplante renal padecen esta enfermedad. La infección de quiste renal es más frecuente en mujeres y suele estar causada por microorganismos entéricos. La TC puede demostrar un quiste complicado sin signos específicos de infección. La gammagrafía con galio es positiva en aproximadamente la mitad de los casos. La gammagrafía con leucocitos marcados con 111 In es más sensible y específica. Los quistes infectados usualmente no responden al tratamiento antibiótico Uno de cada 5 pacientes desarrolla nefrolitiasis. La composición química de los cálculos (generalmente oxalato cálcico o ácido úrico) y la frecuencia de valores subnormales de citrato en la orina y de valores de pH urinario bajos indican la importancia de factores metabólicos, además de factores mecánicos, en la patogenia de esta complicación. En raros casos se ha observado el desarrollo de un carcinoma renal en individuos con riñón poliquístico autosómico dominante. La detección de esta complicación puede ser muy difícil. La poliquistosis hepática generalmente es asintomática, se desarrolla más tarde que la poliquistosis renal y suele ser más grave en las mujeres que en los varones.Sin embargo, hasta los hígados más afectados contienen suficiente parénquima residual para mantener una función hepática normal. Estudios angiográficos utilizando RM detectan aneurismas intracraneales en el 5-10% de los pacientes con poliquistosis renal. La mayoría de estos aneurismas son de tamaño muy pequeño, con un diámetro inferior a 6 mm, y probablemente el riesgo de rotura es muy bajo. No obstante, la incidencia de hemorragia subaracnoidea por rotura de un aneurisma intracraneal en la poliquistosis renal es de 5 a 10 veces superior a la observada en la población general. Otros trastornos asociados a la poliquistosis renal autosómica dominante incluyen anomalías cardiovasculares (prolapso de la válvula mitral, dilatación de la raíz aórtica, aneurismas o disecciones aórticas, válvula aórtica bicúspide y coartación de aorta), divertículos de colon, hernias inguinales y anomalías esqueléticas congénitas. Diagnóstico: El método de elección para el diagnóstico presintomático de la enfermedad es la ecografía que se realizará a partir de los 20 años en todos los sujetos con antecedentes familiares de la enfermedad. Además el análisis genético hace posible el diagnóstico de la enfermedad a cualquier edad en familias con varios individuos afectados en las que se haya podido establecer el ligamiento genético. La TC detecta los quistes sin ninguna dificultad (*) Tratamiento: Está dirigido a la prevención de complicaciones y, si es posible, a retrasar la progresión de la insuficiencia renal. Debe prestarse especial atención a la detección precoz y al tratamiento estricto de la hipertensión, así como a la corrección de otros factores de riesgo cardiovasculares, ya que las complicaciones cardiovasculares constituyen la principal causa de mortalidad. Deben evitarse los deportes de contacto, el abuso de opiáceos o analgésicos, las manipulaciones innecesarias del tracto urinario y el uso inadecuado de diuréticos o la restricción severa de sodio en enfermos con insuficiencia renal y pérdida renal de sodio. En enfermos con insuficiencia renal es aconsejable una restricción proteica moderada. Debido a la dificultad del diagnóstico diferencial entre pielonefritis aguda e infección de un quiste y a la posible presencia de septicemia, el tratamiento inicial es con una combinación de amplio espectro como ampicilina y un aminoglucósido. Cuando se ha aislado el microorganismo, el tratamiento debe modificarse de acuerdo con las sensibilidades in vitro. Los enfermos que no responden al tratamiento inicial probablemente tienen un quiste infectado, en cuyo caso el tratamiento debe modificarse con la inclusión de un agente liposoluble como ciprofloxacino, trimetoprim, cloramfenicol o metronidazol. La descompresión quirúrgica o laparoscópica
de riñones poliquísticos sólo está indicada en enfermos
con dolor renal crónico incapacitante. Los aneurismas
intracraneales con un diámetro superior a 10 mm deben
tratarse quirúrgicamente; aquellos con un diámetro inferior a
6 mm tienen un bajo riesgo de rotura y deben evaluarse
anualmente. No existe acuerdo general acerca del tratamiento |
REFERENCIAS
|
|
|
|
| Monografía revisada el 29 de Noviembre de 2006. Equipo de Redacción de IQB |