SINDROME HEMOFAGOCITICO REACTIVO
|
|
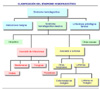
La característica de este síndrome es la proliferación de macrófagos asentados en los tejidos (histiocitos) que además presentan una capacidad fagocítica aumentada sobre las células hematopoyéticas. Este tipo de histiocito puede aparecer en el curso de numerosas enfermedades (*), aunque también hay casos en los que no existe ninguna enfermedad subyacente. Se observa sobre todo en adultos, siendo la proporción de hombre a mujer de 1.5 a 2. Desde el punto de vista clínico se manifiesta por fiebre, citopenias, espleno- y hepatomegalia, adenopatías y alteraciones de la coagulación. Las plaquetas suelen estar por debajo de las 50 x 106/ ml y los leucocitos por debajo de los 2 x 106/ ml, junto a una anemia más o menos intensa. La presencia de hemolisis intramedular queda reflejada en una bilirribina indirecta y una LDH aumentadas. Los histiocitos muestran un aspecto normal, pero son capaces de fagocitar leucocitos, eritrocitos, plaquetas y restos celulares con gran avidez y frecuentemente su tamaño es muy grande debido a la gran cantidad de células fagocitadas (*). La médula ósea es el principal órgano afectado, aunque también pueden encontrarse histiocitos reactivos en el hígado, los ganglios linfáticos y el bazo. No se conoce con exactitud la patogenesis de esta condición. Se cree que algún factor inductor de la fagocitosis o las linfokinas estimuladoras de los macrófagos podrían ser los responsables de la hiperactividad de los histiocitos. Por tanto, el síndrome hemofagocítico reactivo, se puede considerar como una enfermedad autoinmune. El síndrome hemofagocítico es una entidad heterogénea que abarca la proliferación de los histiocitos con hemofagocitosis tanto de forma neoplásica y no neoplásica. Aunque inicialmente el síndrome hemofagocítico fue considerado como un proceso maligno primario llamado histiocitosis maligna o reticulosis medular histiocítica, posteriormente se caracterizó el “síndrome hemofagocítico reactivo” en el que hay proliferación no neoplásica de histiocitos con fagocitosis de células hematopoyéticas, como resultado de la asociación etiológica con bacterias, hongos, parásitos, fármacos y enfermedades malignas (*) Cuando el síndrome hemofagocítico tiene una base hereditaria se denomina como linfohistiocitosis eritrofágica familiar. Las formas reactivas del síndrome hemofagocítico asociadas a enfermedades malignas se dividen en dos grupos:
Se basa en las observaciones de laboratorio. Hay que tener en cuenta que otros procesos como la anemia hemolítica autoinmune, la drepanocitosis y muchos fármacos pueden producir histiocitos con hemofagocitosis. Se considera como positivo el SHR cuando al menos un 2% de todas las células medulares nucleadas son histiocitos hemofagocíticos, acompañadas de fiebre y citopenias (*) El pronóstico depende del tipo de síndrome hemofagocítico considerado. El síndrome hemofagocítico reactivo secundario a infección es una condición benigna y autolimitada, mientras que la histiocitosis maligna es rápidamente progresiva y por lo común fatal, por lo que requiere tratamiento oportuno con quimioterapia. En los síndromes hemofagocíticos asociados a enfermedades malignas, el pronóstico depende de la variedad histológica del tumor. En los enfermos con histiocitosis maligna o histiocitosis eritrofágica familiar, la enfermedad es altamente agresiva, de evolución muy rápida y con poca o ninguna respuesta al tratamiento. TRATAMIENTO No hay ningún tratamiento específico. La medicación debe ir enfocada a tratar la enfermedad subyacente si es que la hay. Se recomienda suspender el tratamiento con inmunosupresores en el caso de que el paciente esté recibiendo corticoides u otros fármacos de este tipo. La condición remite en unos días si la enfermedad subyacente desaparece. REFERENCIAS
|
|
| Monografía revisada 21 de abril de 2011. Equipo de Redacción de IQB | ||
| |