SÍNDROMES MIELODISPLÁSICOS
|
Los síndromes mielodisplásicos constituyen un grupo de trastornos caracterizados por una o más citopenias de la sangre periférica, secundarias a la disfunción de la médula ósea. Los síndromes pueden surgir de novo o de manera secundaria después de haberse aplicado un tratamiento con quimioterapia, radioterapia, o ambos, para otras enfermedades. Históricamente, se han utilizado varios términos para describir estos síndromes tales como anemia refractaria con exceso de mieloblastos, leucemia aguda submieloide, oligoleucemia y síndromes dismielopoyéticos. Estos síndromes de caracterizan por la citopenia de algunas líneas celulares de la sangre, usualmente myeloides/monocitos, eritroides, o megacariocitos, con una médula ósea hipercelular y una hematopoyesis deficiente. Los síndromes mielodisplásicos se clasifican en 5 tipos (*) según el grupo francoamericanobritánico (FAB) de 1982 de acuerdo con criterios morfológicos y, más recientemente, según la OMS en 9 entidades. Aunque la clasificación del FAB tiene un cierto valor pronóstico, varios análisis independientes han propuesto la creación de unos índices pronósticos basados en una serie de variables como son las características y porcentajes de los blastos, la incidencia y grado de las citopenias, la edad, los niveles de lactato-deshidrogenada y las características citogenéticas Los sindromes mielodisplásicos de novo o primarios resultan de varias anormalidades citogenéticas que producen una proliferación neoplásica clonal asociada con una destrucción celular hiperactiva y un aumento de la apoptosis. Así, la coexistencia de crecimiento celular anormal y muerte caracterizan algunas de las mielodisplasias como desordenes hematológicos difíciles de tratar. Los síndromes mielodisplásicos secundarios difieren de los primarios por la existencia de un antecedente citotóxico, por sus características morfológicas, por cursar con mayores anormalidades cromosómicas y evolucionar también, con mayor frecuencia, hacia las leucemias agudas (*). La incidencia de los síndromes mielodisplásicos es de 4/100.000 siendo la edad media de aparición de 70 años, siendo el 85% de los casos mayores de 60 años. |
 |
La anemia es el síntoma más característico de los síndromes mielodisplásicos. En las formas más moderadas, la anemia grave resulta en fatiga, necesidad de transfusiones y sobrecarga de hierro. En los otros subtipos aumenta la pancitopenia, aumento de blastos en la médula ósea y progreso hacia la transformación con resultado de leucemia mieloide aguda. En uno de los subgrupos de síndromes mielodisplásicos se presentan síntomas inmunológicos con artritis, rash y fiebre Dada la heterogeneidad de la enfermedad, el pronóstico de los síndromes mielodisplásicos es muy variable. En algunos pacientes la enfermedad puede seguir un curso benigno durante muchos años, mientras que otros progresan rápidamente hacia la leucemia o mueren debido a una insuficiciencia medular. De acuerdo con el Indice Internacional de Pronóstico (Internactional Prognostic Scoring System, IPSS) los pacientes se clasifican en 4 categorias (*) con unos tiempos de supervivencia de 5.7, 3.5, 1.2 y 0.4 años para cada uno de los grupos, desde el bajo al alto riesgo |
|
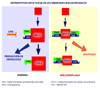
PATOFISIOLOGIA DE LOS SÍNDROMES MIELODISPLASICOS La mielodisplasia se caracteriza por citopenias refractarias de múltiples lineas celulares de la sangre que hacen necesarias transfusiones periódicas, por una aumento excesivo de los riesgos de hemorragia y de infección y por un aumento del potencial de una transformación a leucemia mieloide aguda. La hematopoyesis defectuosa es la consecuencia de una serie de interacciones muy complejas entre los progenitores hematopoyéticos y los multiples factores ambientales que conducen a una apoptosis prematura tanto de los progenitores como de su descendencia. Los progenitores mielodisplásicos de la médula osea muestran una capacidad deficiente para generar células madre pluripotentes y eritroides, maduran más lentamente y responden de manera inadecuada a los factores de crecimiento, a pesar de exhibir unos receptores normales a estos factores. La única excepción es la leucemia mielomonocítica crónica, en la cual las células progenitoras muestran una hipersensibilidad al factor estimulante de las colonias de granulocitos (GM-CSF) lo que se traduce en una monocitosis periférica Los progenitores eritropoyéticos muestran una respuesta defectuosa del factor de transducción STAT-5 y del factor especifico del eritroide GATA-1 cuando son estimulados por la eritropoyetina. Los progenitores eritroides normales contienen y expresan los receptores FAS o CD95 (unos receptores que actúan como ligandos de unas proteínas llamadas, fasL; cuando estos receptores son activados - por ejemplo con el interferon g - se produce la unión fas-fasL, acasionándose la apoptosis celular), pero su densidad es baja, y, por tanto, no está favorecida la vía de la apoptosis. Por el contrario, en las células mielodisplásica, tanto la densidad de los receptores FAS como la concentración del ligando fas están aumentadas, lo que permite la activación autocrina y paracrina de la muerte celular programada (*). La intensidad de la expresión de los receptores FAS está inversamente relacionada con el número de blastos, mientras que la concentración del ligando fas permanece constante: a medida que la enfermedad progresa, las poblaciones de mielobastos que van emergiendo son cada vez más resistentes a la apoptosis inducida por el ligando fas. Adicionalmente, la regulación de los progenitores hematopoyéticos está parcialmente modulada por el factor de necrosis tumoral (TNF-a) y otras citocinas inflamatorias como p.ej la interleukina-1b. En la médula ósea de los pacientes con síndromes mielodisplásicos se observa una superproducción de estas citocinas, citocinas que promueven la apoptosis. En consecuencia, se produce la muerte de los progenitores mielobásticos que no pueden diferenciarse en último término a eritrocitos, con la correspondiente anemia refractaria. La eritropoyesis es un mecanismo homeostatico en el que la reducción del número de eritrocitos produce un efecto retroalimentador sobre la producción de blastos. Se produce entonces un superproducción de blastos: parte de ellos se acumulan en la médula ósea, parte de ellos experimentan apoptosis antes de diferenciarse a eritrocitos y parte de ellos migran y pasan a la sangre. De esta manera, se producen los diferentes estadios de la enfermedad y el aspecto caracteristico de los frotis de medula osea y de sangre cuando hay un exceso de blastos |
|
El diagnóstico Sangre periférica:
Médula ósea
|
|
REFERENCIAS
|
|