ANEMIA DREPANOCITICA [ICD-10: D57] |
|
|
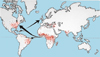
La anemia falciforme o anemia drepanocítica pertenece al grupo de hemoglobinopatías más frecuentes. Es ocasionada por una hemoglobina S, así llamada por su inicial en inglés "sickle" = hoz, debido a la forma que adoptan los eritrocitos cuando disminuye su oxigenación. Descubierta en 1910 en un estudiante con una enfermedad dental, Herrick acuñó el término de "enfermedad falciforme" debido a la forma de los eritrocitos. Sin embargo, tuvieron que pasar 15 años antes de que esta enfermedad fuera considerada como primaria y no debida a una infección u otra condición. En 1927 Hahn y Gillespie demostraron que se podía provocar esta forma saturando de dióxido de carbono una suspensión de eritrocitos, y hacia 1945 Pauling sugirió que la enfermedad se debía a un anormalidad de la molécula de hemoglobina a la que denominó hemoglobina S. Poco después, se evidenció que la hemoglobina S era el resultado de una mutación en el codón 6 del gen beta de la globina en la que la base timina es sustituída por la adenina (GTG--> GAG) lo que ocasiona que el glutámico en b6 de la globina sea sustituído por valina. Como esta sustitución se lleva a cabo en una posición superficial de la molécula de hemoglobina y la carga eléctrica es diferente, la movilidad electroforética de la HbS es menor que la de la hemoglobina normal, pudiéndose ser fácilmente separadaLa anemia falciforme afecta a las personas que heredan dos copias del gen mutado de sus padres portadores. Los padres portadores tienen una probabilidad entre cuatro de tener un hijo enfermo y una probabilidad entre dos de tener un hijo portador no afectado. Los portadores no son afectados pero pueden transmitir la enfermedad Debido a que los eritrocitos portadores de la hemoglobina S son resistentes a la infección por Plasmodium falciforum (responsable del paludismo), la distribución geográfica de esta hemoglobinopatía discurre paralela a las áreas donde ha existido o existe esta enfermedad endémica. La mayor parte de los casos se encuentran en el África tropical donde hasta el 45% de la población es portadora de la mutación. También es frecuente en las áreas con población del color de EE.UU y Centroamérica Se conocen varios genotipos de anemia falciforme, caracterizados todos ellos porque la hemoglobina S constituye al menos la mitad de la hemoglobina presente: la forma más común homocigota para la hemoglobina S o anemia falciforme propiamente dicha, la hemoglobinopatía S/B talasemia (clínicamente indistinguible de la anemia falciforme), la hemoglobinopatía HbSC (doble heterocigota para la HbS y la hemoglobina C) y hemoglobinopatía S con persistencia hereditaria de hemoglobina fetal, síndrome HbS/HbE (un síndrome muy raro con un fenotipo similar al de la HbS/B talasemia y otras combinaciones muy poco frecuentes de hemoglobinopatía S con otras hemoglobina anormales (Angeles, G-Philadelphia, HbO Arab, etc) En los últimos años, la esperanza de vida ha mejorado notablemente estimándose que el 85% de los niños con anemia falciforme sobreviven hasta los 18 años y que los hombres llegan a vivir 42 años y las mujeres hasta los 48 años |
||
|
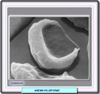
Fisiopatología de la anemia falciforme Como consecuencia de la mutación, cuando la hemoglobina se desoxigena, sufre un proceso espontáneo de polimerización formando un gel cristalino. Cada polímero está formado por 14 haces longitudinales de deoxi-Hb que se disponen formando un cuerpo tactoide, estructura cilíndrica insoluble y rígida. Debido a estos polímeros, se rompe el citoesqueleto del eritrocito, adoptando este la forma característica del drepanocito. Aunque el fenómeno de la falciformación es reversible, entre el 5 y el 50% de los eritrocitos falciformes no consiguen recuperar su forma original, siendo eliminados por el sistema mononuclear fagocítico. Por otra parte, los eritrocitos alterados presentan un gran descenso del volumen corpuscular y un gran aumento de la concentración de hemoglobina. Esto es debido a que la deoxi-Hb induce alteraciones de la membrana eritrocitaria (modificación de la composición y distribución de los fosfolípidos en la bicapa) que se traducen por una profunda deshidratación. Adicionalmente, los drepanocitos exhiben una gran tendencia a adherirse al endotelio vascular favoreciendo la formación de microtrombos y oclusiones vasculares periféricas Es interesante destacar que la hemoglobina S puede interaccionar con otras formas de hemoglobina, en particular con la hemoglobina fetal (HbF). En presencia de esta forma de hemoglobina, se reduce el grado de polimerización de la HbS, lo que explica que la anemia falciforme no se presente nunca durante el período neonatal o en la persistencia hereditaria de la hemoglobina fetal. |
||
Diagnóstico La confirmación diagnóstica de la anemia falciforme o de su carácter portador se lleva a cabo mediante el hemograma (que muestra anemia macro- o microcítica y reticulocitosis), el examen morfológico del frotis que muestra la presencia de numerosos drepanocitos y la electroforesis de las hemoglobinas a pH alcalino que permite determinar el tipo de hemoglobina predominante, y que en el caso de la anemia falciforme homocigota es mayoritario con ausencia total de hemoglobina HgA normal. En el caso de la anemia falciforme con portador heterocigoto las bandas electroforéticas de las HbA y HbS suelen ser de la misma intensidad. Para confirmar el carácter drepanocítico del componente hemoglobínico anormal se lleva a cabo el examen microscópico de los hematíes sometidos a desoxigenación sobre el portaobjetos. |
|||
| En el diagnóstico diferencial están incluídas todas las manifestaciones clínicas y complicaciones de la anemia falciforme | |||
|
La hemoglobinopatía S se presenta en dos formas clínicas: la homocigota, la más frecuente, en la que los pacientes experimentan anemia hemolítica y la heterocigota, generalmente asintomática. La intensidad de la enfermedad, que se manifiesta a partir de los 3 o 4 meses de edad, depende de su coexistencia con otras hemoglobinopatías asociadas y es muy variable de unos pacientes a otros. Se reconocen tres fases evolutivas de la anemia falciforme, cada una de ellas con una sintomatología característica: La fase estacionaria corresponde a los 1 a 4 primeros años de vida y los síntomas son los de un síndrome hemolítico crónico moderado o intenso con una intensa retención esplénica de los eritrocitos y complicaciones vaso-oclusivas. Los niños muestran hiperesplenismo, anemia, palidez cutáneomucosa, ictericia subconjuntival y retraso en el crecimiento. La fase de expresividad aguda se inicia a partir de los 4 años, con agravamiento del cuadro anémico y aparición de diversas manifestaciones clínicas de carácter vasooclusivo, así como infecciones recidivantes, que son las responsable de un elevado porcentaje de muertes. La más frecuente de estas infecciones es la osteomielitis, producidas casi siempre por Salmonellas. Además los pacientes experimentan crisis drepanocíticas muy dolorosas que obedecen a pequeños infartos de la microvasculatura en los miembros superiores e inferiores (dactilitis con síndrome mano-pie). Cuando ocurren accidentes vasooclusivos en vasos grandes, las consecuencias son más graves. Son de destacar los infartos mesentéricos, que cursan con dolores abdominales agudos, la afectación del sistema vascular pulmonar que pueden producir hipertensión pulmonar e insuficiencia respiratoria graves y trombosis de la arteria central de la retina que puede ser la causa de amaurosis. |
|||
|
|
La fase de expresividad crónica se observa solo en los pacientes que han sobrevivido la infancia, siendo muy numerosas las complicaciones que ocasiona. En más del 50% de los pacientes aparecen úlceras maleolares favorecidas por traumatismos e infecciones. Otras complicaciones más o menos frecuentes son las necrosis óseas asépticas, las retinopatías proliferativas muy parecidas a las diabéticas, insuficiencia pulmonar crónica frecuentemente con hipertensión pulmonar, sobrecarga funcional cardíaca y complicaciones renales, en particular, incapacidad para concentrar la orina. |
||
Tratamiento Por el momento, la anemia falciforme no tiene curación y, por lo tanto, los tratamientos son paliativos y preventivos de las complicaciones. Los objetivos del tratamiento son el manejo de las crisis vasooclusivas, el alivio de los dolores crónico, el tratamiento de la anemia hemolítica crónica, la prevención y tratamiento de las infecciones, el tratamiento de las complicaciones en los órganos afectados por la enfermedad, la prevención del ictus y la detección y tratamiento de la hipertensión pulmonar. La aparición de una crisis drepanocítica aguda con dolores intensos y fiebre de 38º o más requiere un tratamiento de urgencia para aliviar el dolor, descartar la posibilidad de una infección y prevenir la deshidratación. Las crisis vasooclusivas son tratadas mediante una hidratación adecuada con fluídos intravenosos (salino y dextrosa al 5%); para el tratamiento del dolor, se recomiendan los opiáceos, siendo la morfina el fármaco de primera elección. El tratamiento de la anemia aguda requiere la práctica de frecuentes transfusiones hasta conseguir valores de hemoglobina del orden de los 100 g/L. Es frecuente recomendar pautas de inmunización preventiva, en particular frente a Streptococcus pneumoniae y Haemophilus influenza. Algunos pediatras aconsejan administrar de forma preventiva penicilina por vía parenteral cada 3 semanas. En los últimos años se han intentado utilizar fármacos antidrepanocíticos, con objeto de estabilizar le membrana del eritrocito, disminuir la tendencia de la deoxi-hemoglobina a la polimerización, aumentar la afinidad de la hemoglobina S hacia el oxígeno y mejorar las propiedades reológicas de los eritrocitos con objeto de evitar las vasooclusiones. Aunque el clotrimazol por vía oral ha demostrado prevenir la deshidratación celular al estabilizar la membrana eritrocitaria inhibiendo el transporte de iones Cl- y K+, solo existen unos pocos datos clínicos en sujetos normales y pacientes voluntarios. Solo la hidroxiurea, un agente alquilante inhibidor de la fase S del ciclo celular empleado en la policitemia vera, está autorizado para su uso en la anemia falciforme. Este fármaco actúa directamente sobre el gen beta de la globina y aumenta los niveles de HbF reduciendo la adhesividad de los eritrocitos y, por tanto, los accidentes vasooclusivos. Sin embargo, la hidroxiurea es muy tóxica y potencialmente carcinogénica. En casos de extrema gravedad, algunos autores consideran el trasplante de médula ósea como una alternativa terapéutica Tratamientos futuros: Terapia génica: tiene por objeto integrar el gen terapéutico en sustitución del gen mutado que codifica la cadena beta de globina en los precursores hematopoyéticos, con objeto de evitar la polimerización de la HbS Tratamiento farmacológico: son varios los fármacos en fase de desarrollo con el propósito de evitar la deshidratación de los eritrocitos. El senicapoc (ICL-17043) es un bloqueante del canal Gardos (este es un canal para potasio calcio-dependiente, más abundante en los drepanocitos) que impediría la salida de potasio y de cloro, preservando el contenido de agua. Este mismo mecanismo de inhibición del canal Gardos es el que explica la eficacia del clotrimazol oral y sobre todo de su metabolito ICA-17043, 10 veces más potente que el clotrimazol, actualmente en fase II (primavera del 2009) Con objeto de evitar la adhesividad de los drepanocitos se está estudiando el RheothRx, un surfactante no iónico que aumenta el flujo sanguíneo al reducir la viscosidad y las fuerzas de fricción. Algunos estudios preliminares parecen demostrar que la administración intravenosa de RheothRx reduce el dolor y la incidencia de accidentes vasooclusivos
|
|||
REFERENCIAS
|
|||
|
|||
|
|||
|
|||
| Monografía revisada el 29 de septiembre de 2016. Equipo de Redacción de IQB | |||

|
|||
|
|
|||