
ANOMALIAS ESTRUCTURALES EN LA ENFERMEDAD DE ALZHEIMER
|
|
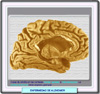
La enfermedad de Alzheimer es una enfermedad neurodegenerativa devastadora, caracterizada clínicamente por un inicio insidioso, un declive progresivo que las funciones cognitivas con un pérdida fatal en último término de las funciones mentales. La enfermedad se caracteriza por lesiones neuropatológicas que se manifiestan como depósitos proteínicos que se localizan preferentemente en el hipocampo y en las áreas parietoremporales de la corteza cerebral (*). Esta lesiones consisten en placas neuríticas compuestas por depósitos extracelulares de beta-amiloide (placas de b-amiloide) y por ovillos intereuronales formados por neurofibrillas consistentes en filamente enrrollados de la proteína tau citoesquelética. Estos dos procesos degenerativos, se potencializan y provocan una degeneración de las células nerviosas implicadas en la memoria y las funciones cognitivas superiores La enfermedad de Alzheimer se caracteriza por una serie de anomalías cerebrales que afectan alguna regiones específicas en particular:
|
|
|
El beta-amiloide, descubierto en 1984 por Glenner et cols., es un péptido de 39 a 42 aminoácidos que se origina a partir de la llama proteína precursora del amiloide (APP) por acción de peptidadas llamadas secretasas. Existen dos tipos más importantes de b-amiloide, denominados b-amiloide 1-40 y b-amiloide 1-42 según el número de aminoácidos presentes. La APP es un proteína transmembrana, que tiene 770 aminácidos y que se encuentra en diferentes tipos de células. En su porción extracelular, la APP posee varias regiones con actividad neurotrófica (*). La proteína precursora del amiloide es miembro de una familia que comprendo dos proteínas semejantes a ella, la APLP1 y la APLP2. La APP está presente en dendritas, cuerpos celulares y axones de neuronas y es su función neuronal es desconocida. La APP es sintetizada en el retículo endoplásmico rugoso, glicosilada en aparato de Golgi y liberada en la membrana como una proteína transmembrana, quedando la porción 613-671 que contiene el b-amiloide parcialmente incluído en la membrana (*). El gen de la APP se encuentra situado en el cromosoma 21. Varios investigadores han detectado en varias familias de enfermos en los que la enfermedad se declara prematuramente, mutaciones en el gen de la APP situadas en diferentes partes del mismo: así, una familia sueca se ha encontrado una doble mutación en los codones 670 y 671, y en otras familias, se detectado una mutación en el codón 717 (*). En estas familias, solo las personas que muestran estas mutaciones padecen la enfermedad de Alzheimer, lo que demuestra sin ningún género de dudas que existe una relación entre el metabolismo de la APP y en la enfermedad de Alzheimer. La APP puede experimentar un metabolismo diverso según que genere como producto final b-amiloide u otros fragmentos.
|
|
|
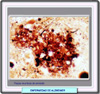
Los depósitos de amiloide están formados en su mayor parte por b-amiloide y otros constituyentes presentes en menor cantidad, en particular la apolipoproteína E (ApoE) y distintos tipos de proteoglicano. La ApoE se une firmemente al péptido amiloide b, lo que favorece la formación de la fibras amiloides. De hecho, muchos autores consideran la ApoE como un cofactor de la amiloidegénesis. La apolipoproteína E forma parte de los quilomicrones y de las lipoproteínas de muy baja densidad (VLDL) presentes en el plasma. En el hígado, la ApoE se une a un receptor específico, siendo esencial para el catabolismo de los ácidos grasos. La ApoE es un polipéptido que contiene 317 aminoácidos y el gen que la codifica se localiza en el cromosoma 19, asociado a los genes ApoC1 y ApoC2. Se conocen 3 isoformos de esta proteína denominados E2, E3, yE4, de acuerdo con su posición relativa después de un enfoque isoléctrico. Cada una de ellas, está codificada por los correspondiente alelos e2, e3 y e4. La ApoE más frecuente es la E3 que está presente en el 40-90% de la población (*). Las ApoE tiene la misión de reparar las neuronas aportando los lípidos necesarios. En este sentido, la más eficaz es la ApoE2. La mayor expresión de la ApoE4 suele ir asociada a una serie de demencias como la enfermedad de Alzheimer esporádica, la demencia vascular, la demencia con cuerpos de Lewy y la enfermedad bulbar de las neuronas motoras. Sin embargo, no todos los individuos que muestran un exceso de ApoE4 desarrollan demencia. Los depósitos de amiloide aparecen ampliamente distribuídos en la materia gris cortical observándose gránulos difusos tioflavina positivos (*). En muchos casos, el b-amiloide se encuentra rodeando las arteriolas corticales y leptomeníngeas (*) El amiloide
parece lesionar progresivamente las neuronas, en particular las implicadas
en las funciones intelectuales. |
|
|
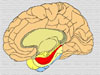
El segundo proceso degenerativo que tiene lugar en la enfermedad de Alzheimer (y en otras enfermedades degenerativas del sistema nervioso) es la formación de ovillos de neurofibrillas que se inicia en la región del hipocampo (*) en la que reside la función de la gestión de la memoria. En este proceso están implicadas la proteínas tau, por lo que muchas veces las enfermedades en las que se observa la formación de neurofibrillas se denominan taupatías. En 1963, se descubría que los ovillos de neurofibrillas eran estructuras filamentosas acumuladas en el citoplasma de las neuronas degeneradas, filamentos que fueron denominados "parejas de filamentos helicoidales" o PHFs. Estos filamentos están formados por microtúbulos citoesqueléticos asociados a las llamadas proteínas tau, que pertenecen a la famila de la "Proteínas asociadas a los microtúbulos" o MAP. En la enfermedad de Alzheimer, las neuronas que muestran estos ovillos de neurofibrillas son sobre todo células piramidales que tienen una forma característica (*) Desde su descubrimiento, las proteínas tau han sido extensamente estudiadas por su implicación en la enfermedad de Alzheimer y otros tipos de demencia. En el cerebro humano, las proteínas tau se presentan como seis isoformas de 352 a 441 residuos de aminácidos que provienen todas ellas de la expresión de un único gen tau localizado en el brazo largo del cromosoma 17. Se conocen perfectamente todas las propiedades físico-químicas de esta proteínas y como interaccionan con los microtúbulos neuronales estabílizándolos. Las proteínas tau se caracterizan por fosforilizarse fácilmente, permitiendo esta reacción la movilidad de las mismas dentro de los axones de las neuronas en direccción centrífuga. En la enfermedad de Alzheimer y otras tauopatías se produce un fenómeno de hiperfosforilización y/o de fosforilización anormal que en definitiva, es el responsable de la formación de los complejos proteínas tau-PHFs (*). En la actualidad, los esfuerzos de los investigadores se encaminan a la idenficación de las proteína-quinasas implicadas en esta fosforilización anormal. El conocimiento de un estructura permitiría eventualmente, el diseño de un inhibidor enzimático que frenase o eliminase las proteínas tau anormales y por lo tanto la formación de ovillos de neurofibrillas. Algunos autores opinan que la fosforilización anormal de las proteínas tau no es el único factor implicado en la formación de los ovillos de neurofibrillas y que podrían intervenir otras moléculas como la Apolipoproteína E o el péptido b-amiloide con lo que los dos procesos más importantes de la enfermedad de Alzheimer estarían interrelacionados. La patología tau siempre precede a la aparición de b-amiloide En resumen, los dos procesos anatomopatógicos implicados en la enfermedad de Alzheimer podrían estar interrelacionados y, en cualquier caso, conducen a una degeneración y disfunción neuronal (*) |






 |

  |
 |