|
ENFERMEDAD
DE NIEMANN-PICK [ICD-10: E75.2]
|
|
|
|
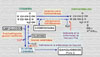
La enfermedad de Niemann-Pick es un desorden en el que se produce una acumulación tisular de esfingomielina, principalmente en los histiocitos, debida a un déficit de esfingomielinasa. Su herencia es autosómica recesiva. Se presenta sobre todo en niños pequeños, pero también existe una forma del adulto esencialmente visceral. La esfingomielina es una enzima lisosomal codificada por un gen localizado en el cromosoma 11p15.1-p15.4. (*) Clínicamente cursa con trastornos neurológicos y hepatoesplenomegalia. También puede haber adenopatías, erupción xantomatosa, infiltrados pulmonares difusos y manchas de color rojo-cereza en la retina (*) . Según el grado evolutivo y la presencia o ausencia de afección del sistema nervioso se consideran varias formas de la enfermedad, denominados tipos A, B, C, D
Los signos clínicos neurológicos que presentan estos pacientes son paresia de la mirada vertical de tipo supranuclear, ataxia, deterioro intelectual y trastornos extrapiramidales (distonía y coreoatetosis) (*) Los pacientes pueden presentar diversos grados de citopenia y la presencia de linfocitos y monocitos vacuolados. El tipo A suele ser fatal en la primera infancia, mientras que los niños con enfermedad del tipo B suelen sobrevivir aunque con hepatoesplenomegalia.
|
||
|
|
Además de la historia y examen clínico, las células de Niemann-Pick son de gran tamaño, con un núcleo excéntrico y un citoplasma abundante y pálido, finamente vacuolado y de aspecto espumoso (foam cells) (*). En las secciones teñidas con hematoxilina-eosina algunas células contienen un pigmento amarillo o amarillento-marrón que es material ceroide o su equivalente, la lipofuscina. En la tinción de Giemsa, algunos de los macrófagos pueden contener gránulos en el citoplasma de color azul o azul-verdoso, que les confieren la denominación de histiocitos azul marino. Al MET, las inclusiones lipídicas ofrecen en su periferia una disposición lamelar concéntrica (*) . Dichas células se observan principalmente en el hígado, bazo, ganglios linfáticos, medula ósea, cerebro y, ocasionalmente, en la piel. Su presencia es indiciaria de la enfermedad de Niemann-Pick pero no diagnóstica, puesto que en otras tesaurismosis también pueden verse histiocitos de aspecto espumoso. Para el diagnóstico se requiere la demostración química de la acumulación de esfingomielina o del déficit de la enzima. Tratamiento Se ha conseguido la normalización de la actividad de esfingomielinasa en dos enfermos tras el trasplante de medula ósea. Los valores de esfingomielina plasmática se normalizaron en ambos niños durante más de un año, aunque la esplenomegalia y los trastornos neurológicos no se modificaron. |
||
| El miglustat puede mejorar o estabilizar las manifestaciones neurológicas en pacientes pediátricos con las formas-finales infantiles y juveniles de aparición de la enfermedad de Neumann-Pick tipo C . Entre los pacientes-infantiles con aparición temprana de la enfermedad, el plazo más breve entre el inicio de la enfermedad neurológica y la iniciación del tratamiento con miglustat se asoció con un mejor resultado terapéutico inicial en un paciente. Sin e,mbargo, el miglustat no parece modificar el curso general de la enfermedad en este subgrupo | |||
|
REFERENCIAS
|
|||
|
|||
|
|||
| Monografía revisada el 20 de septiembre de 2014. Equipo de Redacción de IQB | |||
|
|
|||