|
|||
|
LEUCEMIA
PROMIELOCÍTICA AGUDA [ICD-10: M9866/3) C92.4 ] |
|
|
La leucemia
promielocítica aguda, también conocida como leucemia mieloide-3
(o M3 de la clasificación FAB), se caracteriza por un predominio
de promielocitos malignos que muestran una translocación recíproca
entre los brazos largos de los cromosomas 15 y 17, t(15;17)(q22;q11.2-q12).
Como consecuencia de esta translocación se produce una fusión
del gen situado en el locus 15q22 (que recibe el nombre de gen PML -
iniciales de ProMyelocytic Leukaemia-)
con el gen para el ácido retinoico (RARa),
este último localizado en 17q12-21. De esta manera, se forma
un gen híbrido PML-RARa, que está
presente en la mayoría de los casos de leucemia promielocítica
aguda y su recíproco, RARa-PML que
se presenta en el 60% de los casos La expresión del gen PML-RARa produce una proteína que contiene los dominios de dimerización y de unión al DNA del gen PML nativo, y los dominios de unión al DNA y a otros ligandos del receptor al RARa. Esta proteína muestra un efecto dramático sobre la arquitectura nuclear del promielocito, produciendo la ruptura de los llamados cuerpos nucleares de la PML que son componentes estructurales criticos, haciendo que la maduración del promielocito quede bloqueada en el estadio de promielocito y no pueda seguir la cadena de diferenciación mieloide. Además, la desorganización de los cuerpos nucleares juega un papel fundamental en la patogenesis de la leucemia promielocitica aguda al inhibir la apoptosis celular. La presencia de receptores al ácido a-trans-retinoico en las proteínas híbridas explica que este fármaco, administrado terapeúticamente, induzca el paso de promielocito a mielocito, ocasionando un gran número de remisiones. Uno de los más importantes avances en el conocimiento de la patogénesis y del tratamiento de la leucemia promielocítica aguda ha sido el aclarar la paradoja aparente de que la destrucción del RARa provocada por la translocación APL-RARa bloquea la diferenciación del promielocito, mientras que la administración de un ligando a dicho RARa (ácido retinoico en forma de ácido t-retinoico o tretinoína) permite la diferenciación de las células leucémicas. El receptor al ácido retinoico posee varias regiones funcionales que regulan la unión al DNA, la interacción con varios co-activadores y co-represores y los receptores para varios ligandos, entre ellos el ácido retinoico. El llamado complejo de represión trancripcional modifica la cromatina en las células normales impidiendo la transcripción, pero son suficientes concentraciones fisiológicas de ácido retinoico para desplazar dicho complejo.(*) En el caso de la APL, los promielocitos leucémicos muestran una mayor cantidad de complejo de represión, debido a la expresión del receptor procedente de la translocación. Por lo tanto, son necesarias concentraciones farmacológicas de ácido retinoico para permitir la activación de la transcripción (*) Este mecanismo también explica la resistencia al ácido t-retinoico de la leucemia promielocítica producida por la translocación t(11;17)(q23;q12-21) entre el RARa y el gen PLZF. En este caso, la proteína quimérica contiene en el dominio correspondiente al PLZF un lugar de unión para un complejo de represión adicional. La unión del ácido retinoico al receptor RARa-PLZF produce, como antes, el desplazamiento del complejo de represión unido a la parte RARa pero se sigue manteniendo el segundo complejo en la parte PLZF. La tricostatina A, un inhibidor de la HDAC (histona desacetilasa) restaura la actividad transcripcional inducida por el ácido retinoico (*) La leucemia promielocítica aguda fué identificada en 1957 por Hillestad y descrita con más detalle en 1958. Sin embargo, sólo a partir de 1970 se descubrieron los orígenes genéticos de la enfermedad. En 1990-91 los dos genes implicados en la translocación fueron clonados. La APL constituye aproximadamente el 5-10% de todos los casos de leucemia mieloide, aumentando este porcentaje en los países mediterráneos (15%) y en America Latina (30%). Esta leucemia se diagnostica sobre todo entre los 15 y 60 años y afecta por igual a hombres y mujeres.
|
|
|
|
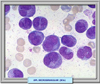
CARACTERISTICAS MORFOLÓGICAS E INMUNOLÓGICAS Se conocen dos variantes morfológicas de la leucemia promielocítica aguda, la APL hipergranular (*) que representa la mayor parte de los casos y la variante microgranular denominada M3v que supone el 15-20% de los casos (*) La forma clásica se caracteriza por la presencia de promielocitos patológicos con abundante coloración azurófila. Los núcleos tienen una forma irregular, presentando escotaduras o lobulaciones (*). Es característica la presencia de abundantes cuerpos de Auer formando astillas apiladas. En muchas ocasiones, la simple observación de los mismos con el microscopio óptico es suficiente para el diagnóstico. La variante microgranular presenta las mismas caracteristicas de la forma de los núcleos pero los gránulos son muy pequeños y no se pueden siempre ver con el microscopio óptico. Esta variante suele presentarse acompañada de hiperleucocitosis. Ambos tipos de APL son fuertemente positivas a las tinciones con myeloperoxidasa, negro de Sudán y cloroacetato-esterasa. Se han desarrollado anticuerpos monoclonales que se fijan específicamente al receptor APL-RARa, que se utilizan para el diagnóstico Desde el punto de vista inmunofenotípico los promielocitos en la APL muestran las características siguientes:
|
|
|
Una de las principales características clínicas en el 80% de los pacientes es la presencia de un síndrome hemorrágico grave. A pesar de los avances el tratamiento de los desórdenes de la coagulación, las hemorragias cerebrales letales siguen siendo la causa de la mayor mortalidad durante la fase de inducción. Aunque inicialmente este síndrome se definía como una coagulación intravascular diseminada, los hallazgos de laboratorio (aumento de los productos de degradación de la fibrina y fibrinógeno, reducción en el número de plaquetas y niveles del inhibidor a2 de la plasmina) apuntan a un proceso fibrinolítico-proteolítico alterado. El reciente hallazgo de que los promielocitos en la APL expresan anexina II (un receptor de membrana para el plasminógeno y para el t-PA) lo que se traduce en una activación del plasminógeno a plasmina, confirma una fibrinolisis anormal con la correspondiente diátesis hemorrágica. Otras características clínicas son la fiebre (15-30% de los casos) y con menor frecuencia, leucocitosis.También son frecuentes los síntomas asociados a anemia y trombocitopenia (*)
|
||
|
|
||
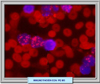 |
Además de la morfología convencional, se dispone de varias técnicas para confirmar la sospecha de una leucemia promielocítica aguda. El método más rápido para la detección de una leucemia promielocítica aguda RARa-PLM positiva es un ensayo de inmunofluorescencia utilizando anticuerpos monoclonales en el que se observa formaciones microparticuladas nucleares características (*) La técnica de RT-PCR (reacción de la polimerasa en cadena con transcriptasa reversa) es esencial para documentar la presencia de genes de fusión PLM-RARa y tratar a los pacientes con los regímenes menos intensos a base de ácido t-retinoico y antraciclinas. También es importante para detectar la mínima enfermedad residual. Otros métodos para la confirmación de una APL son el análisis citogenético, la hibridación fluorescente in situ y la hibridación de Southern.
|
|
|
|
Se han
llevado a cabo en los últimos años varios estudios multicéntricos
utilizando varias combinaciones de ácido trans-retinoico y quimioterapia,
y trióxido de arsénico. Los resultados de estos estudios
indican que el uso concomitante de ácido trans-retinoico (*)
e idarrubicina es superior al uso secuencial, tanto en lo que se refiere
a los efectos antileucémicos como a un mejor control del síndrome
de diferenciación de la APL (también llamado síndrome
RA-APL - ácido retinoico-leucemia promielocítica aguda-
o síndrome del ácido retinoico o síndrome de diferenciación
en la APL) Varios investigadores han demostrado que la probabilidad de una recaída después de la remisión de una leucemia promielocítica aguda está directamente relacionada con el número de células residuales que contengan el gen translocado APL-RARa. Utilizando la técnica RT-PCR que permite detectar una célula leucémica entre 10.000 células normales, se ha demostrado que un estado APL-RARa negativo, está asociado a una mayor supervivencia y una mayor probabilidad de cura total. Estas observaciones han llevado al concepto de remisión molecular (es decir negatividad en el test con RT-PCR para el ALP-RARa) y de recaída molecular (momento en el que el test se hace positivo, sin evidencia hematológica de enfermedad). Estudios realizados en varios países han puesto de manifiesto las ventajas de iniciar un tratamiento tan pronto se detecta una recaída molecular, no solo por el mayor porcentaje de supervivencias en comparación con un tratamiento iniciado al observarse una recaída clínica, sino también por el hecho de que los pacientes tratados muy precozmente muestran una menor incidencia de fatalidades asociadas a problemas hemorrágicos. En caso de recaída después de un tratamiento con ácido retinoico, los pacientes pueden ser tratados con un nuevo ciclo de este fármaco con bastante probabilidades de éxito. Sin embargo, dado que las remisiones no son permanentes, se requiere un intenso tratamiento de consolidación. Estos tratamientos suelen consistir en pequeñas dosis de citarabina, interferón o combinaciones de quimioterápicos (etoposide + mitoxantrona + citarabina; 6-tioguanina + citarabina + daunorrubicina + prednisona; 6-mercaptopurina + vincristina + metotrexato + prednisona; daunorrubicina + vincristina + citarabina + prednisona, etc.) La administración
i.v. de trióxido de arsénico en dosis de 0.15 mg/kg/día
tanto durante la fase de inducción como durante la fase de consolidación
en leucemias promielocíticas agudas refractarias al tratamiento
estándar o en recaídas de la APL ocasiona resultados bastante
satisfactorios: en los estudios realizados, el 87% de los pacientes
tratados con el trióxido de arsénico alcanzaron la remisión
completa Cuando estos tratamientos fracasan después de > remisiones, la fase de consolidación suele consistir en un transplante de médula ósea alogénica o autóloga. Aunque bastante frecuentes, los trasplantes de médula presentan los problemas de la alta toxicidad de la supresión medular y la poca disponibilidad de donantes adecuados. En la actualidad se están realizando estudios clínicos con una nueva formulación del ácido trans-retinoico encapsulado en liposomas para su administracion i.v. y con el gemtuzumab ozamicina (*), un anticuerpo monoclonal anti CD33, que después de fijarse al receptor para CD33 es internalizado en la célula leucémica, liberándose un derivado del antibiotico caliqueamicina que es tóxico para la célula. También se encuentra en fase clínica un nuevo retinoide síntético (CD437) que sería activo en las leucemias resistentes al ácido trans-retinoico (*)
|
|
|
REFERENCIAS
|
||
|
||
|
||
| Monografía revisada el 28 de Mayo de 2013. Equipo de Redacción de IQB | ||

|
||
|
|